
La Sindrome di Ehlers-Danlos (EDS) comprende un gruppo eterogeneo di malattie ereditarie rare che coinvolgono, in modo particolare, il tessuto connettivo. Iperelasticità cutanea, ipermobilità articolare e fragilità di vasi ed organi interni costituiscono la triade clinica più rappresentativa dell’EDS. La prima descrizione della malattia risale al 1892, mentre il nome fa riferimento ad i primi due studiosi che stilarono un elenco delle caratteristiche tipiche di tutte le forme riconosciute della sindrome: Edward Ehlers, un dermatologo danese, e Henry Alexander Danlos, un medico francese esperto in chimica e disordini dei tessuti.
L’EDS nella popolazione generale ha un’incidenza media stimata tra 1/5.000-20.000, potrebbe essere sottovalutata a causa della variabilità clinica. La maggior parte dei pazienti è di sesso femminile. Esordisce a tutte le età, ma si identifica con più difficoltà nei bambini piccoli a causa della maggiore lassità articolare a quella età. La sindrome di Ehlers-Danlos nelle varianti Classica ed Ipermobile sono le forme più frequente di EDS.

Le cause
Le alterazioni a carico del tessuto connettivo che si manifestano nella sindrome di Ehlers-Danlos sono dovute a modificazioni delle proteine che costituiscono il tessuto, prima fra tutte il collagene, della quale esistono oltre 30 tipi differenti. Il collagene è il materiale costitutivo di gran parte dei tessuti e degli organi del nostro corpo, come tendini, legamenti e cartilagine. Le conseguenze di quest’alterazione possono causare problemi in varie parti del corpo. Le modalità di trasmissione della EDS sono diverse: autosomica dominante, autosomica recessiva, recessiva legata all’X. La maggior parte delle forme, fatta eccezione per il tipo cifoscoliotico ed il tipo dermatosparassico, vede una trasmissione autosomica dominante.
Sintomi e segni frequenti nelle Sindromi di Ehlers Danlos:
- La cute è elastica, soffice e vellutata, risulta particolarmente fragile con formazione di lacerazioni e di ecchimosi anche per traumi lievi;
- Le cicatrici guariscono più lentamente con formazione di cicatrici atrofiche, allargate;
- Le articolazioni sono instabili con frequenti sublussazioni;
- Dolore articolare, muscolo-scheletrico precoce, cronico e debilitante;
- Ipotonia muscolare;
- Fragilità tissutale (rottura di arterie, intestino, utero soprattutto nel tipo vascolare);
- Scoliosi;
- Prolasso mitralico.
Tipi e classificazione
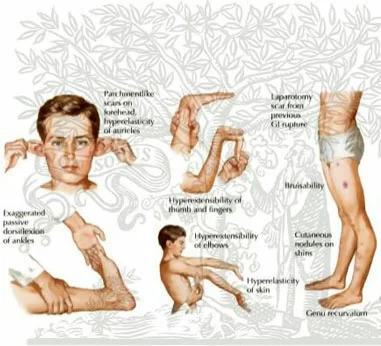
La sindrome di EDS presenta forme particolarmente eterogenee, caratteristica che rende spesso complessa l’identificazione della patologia. Non esistono sintomi universali, poiché anche ciascun paziente rappresenta spesso una patologia a sé stante per espressività, tipologia e gravità della sintomatologia. Le forme più comuni sono la classica e l’ipermobile, mentre la più grave è quella vascolare. La differenziazione tra le forme non è sempre netta e definita, infatti possono essere presenti sintomi sovrapposti tra le varie forme.
L’EDS è attualmente suddivisa in tredici tipi, secondo la Classificazione Internazionale 2017:
- Classico: trasmissione autosomica dominante, prevalenza 1:20.000-40.000;
- Simil classico: trasmissione autosomica recessiva;
- Cardiaco-valvolare: trasmissione autosomica recessiva;
- Vascolare: trasmissione autosomica dominante prevalenza 1:50.000-250.000;
- Ipermobile: trasmissione autosomica dominante, prevalenza 1: 5.000-20.000;
- Artrocalasico: trasmissione autosomica dominante, prevalenza molto rara ad oggi sono riportati circa 30 casi;
- Dermatosparassi: trasmissione autosomica recessiva, molto rara, ad oggi sono riportati circa 10 casi;
- Cifoscoliotico: trasmissione autosomica recessiva, prevalenza 1:100.000, 9;
- Brittle cornea (Cornea Fragile): trasmissione autosomica recessiva;
- Spondilodisplastico: trasmissione autosomica recessiva;
- Muscolocontratturale: trasmissione autosomica recessiva;
- Miopatico: trasmissione autosomica dominante o recessiva;
- Periodontale: trasmissione autosomica dominante.
Classico (cEDS)
I sintomi principali sono:
- ipersensibilità della cute, che si presenta inoltre vellutata, traslucida e sottile, con propensione alla formazione di lividi anche in seguito a lievi traumi;
- cicatrici estese e caratteristiche, cosiddette “a carta di sigaretta”;
- ipotonia muscolare;
- ipermobilità e lassità articolare;
- piede piatto;
- dolore;
- ritardo nello sviluppo motorio.
Ha un’ ereditarietà di tipo autosomico dominante ed è dovuta ad un difetto nel gene COLA541, COLA5A2.
Tipo simil classico ( clEDS)
I sintomi principali sono:
- pelle iperestensibile, fragile e vellutata;
- ipermobilità articolare;
- Ecchimosi spontanee.
- Trasmissione autosomica recessiva.
Tipo cardiaco-valvolare (cvEDS)
I sintomi principali sono:
- Patologie cardiovalvolari (valvola aortica e mitralica);
- Coinvolgimento cutaneo: iperelasticità, cicatrici atrofiche, pelle sottile, fragilità;
- Ipermobilità articolare.
Trasmissione autosomica recessiva.
Tipo ipermobile (hEDS)
La sindrome di Ehlers-Danlos tipo ipermobile (H-EDS) è la forma più frequente di EDS, ed è caratterizzata da:
- lussazioni ed instabilità articolare evidente;
- cute moderatamente iperestensibile;
- scarso trofismo muscolare;
- dolore articolare cronico;
- propensione alla formazione di ecchimosi.
Possono presentarsi anche disturbi cardiaci come il prolasso della valvola mitralica. Ha un’ereditarietà ti tipo autosomico dominante e la causa di questa forma non è tutt’ora ben conosciuta.
Tipo vascolare (vEDS)
I segnali caratteristici della presenza della sindrome sono:
- fragilità delle pareti dei vasi sanguigni (in particolare le arterie), che risultano ben visibili attraverso la cute traslucida e che spesso vanno incontro a rottura;
- facile insorgenza di ematomi, anche per minimi traumi;
- aspetto caratteristico del volto;
- cute sottile ed iperestensibile;
- ipermobilità articolare (solitamente lieve).
Ha un’ereditarietà di tipo autosomico dominante ed è dovuta ad un difetto nel gene COL3A1.
Tipo cifoscoliotico (kEDS)
I pazienti affetti da questa rara forma manifestano:
- lassità articolare generalizzata;
- grave ipotonia muscolare congenita, in particolare in età puberale;
- fragilità della sclera oculare;
- perdita della capacità di deambulazione.
Sono possibili anche fragilità di tessuti, vasi e lividi. Presenta un’ereditarietà di tipo autosomico recessivo.
Tipo artrocalasia (aEDS)
Tipo molto raro, I sintomi principali sono:
- displasia congenita bilaterale delle anche
- grave lassità delle articolazioni;
- lussazioni frequenti;
- ipotonia muscolare;
- scoliosi;
- bassa statura.
Ha un’ereditarietà di tipo autosomico dominante ed è dovuta ad un difetto nel gene COL1A1, COL1A2.
Tipo dermatosparassi (dEDS)
Le caratteristiche principali di questa forma sono:
- Caratteristiche craniofacciali evidenti alla nascita
- fragilità cutanea grave;
- cute iperestensibile che si presenta molle, lassa e soffice;
- ernie ombelicali o inguinali di grandi dimensioni
- Ritardo di crescita postnatale
- Piedi, mani e arti corti.
Presenta un’ereditarietà di tipo autosomico recessivo.
Tipo Bryttle Cornea
Le caratteristiche principali di questa forma sono:
- Cornea sottile;
- Esordio precoce di cheratocono e cheratoglobo;
- sclere blu.
Presenta un’ereditarietà di tipo autosomico recessivo.
Tipo Spondilodisplastico (spEDS)
Le caratteristiche principali di questa forma sono:
- Bassa statura;
- Ipotonia muscolare;
- Arti inferiori ad arco.
Presenta un’ereditarietà di tipo autosomico recessivo.
Tipo Muscolocontratturale (mcEDS)
Le caratteristiche principali di questa forma sono:
- Contratture congenite multiple;
- Piede equino-varo;
- Facies tipica alla nascita (ipertelorismo, sclere blu, naso piccolo…);
- Iperelasticità cutanea, pelle vellutata e traslucida e fragile.
Presenta un’ereditarietà di tipo autosomico recessivo.
Tipo Miopatico (mEDS)
Le caratteristiche principali di questa forma sono:
- Ipotonia muscolare congenita;
- Contratture delle articolazioni prossimali (gomiti, ginocchia e anche);
- Ipermobilità delle articolazioni distali.
Tipo Periodontite (pEDS).
Le caratteristiche principali di questa forma sono:
- Grave e intrattabile periodontite a insorgenza precoce;
- Placche di tessuto cicatriziale nella zona pretibiale;
- Iperplasia delle gengive con ipercheratosi variabile.
Presenta un’ereditarietà di tipo autosomico dominante
Gioia di Biagio
Gioia Di Biagio è una scrittrice, musicista e performer italiana colpita dall’Ehlers-Danlos. Autrice del libro “Come oro nelle crepe”- un racconto autobiografico della propria condizione -, Gioia ci ha raccontato quanto questa malattia sia “diversa da persona a persona”. Le caratteristiche peculiari sono evidenti nella pelle “elastica, delicata, fragile, come seta” e “appena batte contro qualcosa, si lacera”.
Diagnosi
Una corretta diagnosi è determinante, anche se spesso non immediata: la malattia è rara e un medico di famiglia può ritenersi fortunato se vede un caso di EDS durante la sua vita professionale. Per questa ragione, in caso di sospetto di malattia, è importante rivolgersi ad uno specialista. Confermare la diagnosi per l’EDS è una procedura complessa, in alcuni casi non è possibile avere un riscontro diretto mediante test, come per il tipo “Ipermobile”, per il quale non esiste alcun esame specifico. Il metodo di valutazione più efficace rimane forse la storia clinica familiare del paziente. Alcuni dei test utilizzati per la diagnosi sono:
- test diagnostici prenatali, utili per rilevare mutazioni di geni associati nel feto se un parente stretto presenta la malattia. Il tipo “Cifoscoliosi” e la forma “Vascolare” possono essere rilevati prelevando un campione di liquido amniotico (amniocentesi) sul quale si valutano i livelli di attività enzimatica;
- test genetici, per rilevare le alterazioni a carico dei geni coinvolti, utili in particolare nei tipi “vascolare” e “artroclasico”;
test delle urine, utile in particolare per identificare il tipo “Cifoscoliosi” (il test misura il livello di un enzima prodotto dalla mutazione di un gene); - biopsia (analisi istopatologica), che consiste nel prelevare un campione di tessuto sul quale possono essere rilevate eventuali anormalità a carico delle fibre di collagene. Il tipo “vascolare” viene in genere riconosciuto tramite biopsia della pelle;
- ecodoppler ed ecocardio, grazie ai quali è possibile evidenziare anomalie valvolari (il prolasso della valvola mitralica può essere facilmente associato ai tipi “ipermobile” e “classico”.
Trattamento e terapia della Sindrome di Ehlers-Danlos
La terapia della sindrome di Ehlers-Danlos è piuttosto individuale, considerata la differente sintomatologia delle varie tipologie e la variabilità da paziente a paziente che può esistere anche all’interno della medesima forma. Ad oggi non esistono ancora cure efficaci che possano agire alla base del problema (ad esempio sui difetti del collagene). Molti medici sconsigliano ai pazienti affetti da EDS di praticare sport che possano comportare complicanze per tendini e cute e di effettuare sollevamenti ripetuti e trasporto di oggetti pesanti, pur sottolineando l’importanza di mantenere un buon trofismo muscolare così da stabilizzare il più possibile le articolazioni. E’ particolarmente importante proteggere la cute e le articolazioni da possibili traumi, ad esempio attraverso l’uso di tutori e protezioni, da utilizzare durante alcune attività sportive.
Il valore degli interventi ortopedici
Gli interventi ortopedici sono da valutare con grande attenzione perché non sempre sono risolutivi. Sembra che la vitamina C abbia degli effetti benefici anche se non esiste ancora una casistica sufficiente in grado di dimostrarlo. I pazienti affetti da tipo “Vascolare”, essendo a rischio per aneurisma arterioso e possibile rottura dello stesso, è bene che si sottopongano ad ecocardio frequenti. Le lussazioni sono talmente frequenti che in genere i pazienti imparano a ridurle da soli o cercano di limitarle con l’uso di appositi tutori.
Il dolore cronico (spesso di origine muscoloscheletrica) è uno dei maggiori determinanti per il deterioramento della qualità di vita nella SED. La cronicizzazione del dolore e la resistenza al trattamento sono le componenti più rilevanti che influenzano la prognosi. Il dolore, può essere trattato con farmaci antidolorifici specifici, ma sempre sotto controllo del medico curante. Il trattamento fisioterapico consente di aumentare l’efficienza dei muscoli e dei tendini ed il coordinamento neuromuscolare e quindi migliora la stabilità delle articolazioni e contribuisce a diminuire il dolore. Un programma di trattamento adeguato dovrebbe altresì comprendere terapia occupazionale e terapia cognitivo-comportamentale.
La Sindrome di Ehlers-Danlos nei bambini
Il bambino affetto da EDS può praticare attività sportive come il nuoto, che non mobilizzano eccessivamente le articolazioni; sono da evitare invece tutti gli sport che prevedano contatti o carichi eccessivi, come danza o ginnastica artistica. Per limitare i danni futuri è consigliabile cercare di evitare che il bambino esibisca in famiglia o con gli amici la sua ipermobilità articolare, poiché ogni iperestensione rappresenta un microtrauma per l’articolazione.
Pazienti in cui esiste un valido sospetto di EDS o siano stati diagnosticati come affetti da EDS, hanno diritto ad eseguire qualunque prestazione sanitaria in regime di esenzione totale dal ticket sanitario. Tale esenzione avviene tramite codice specifico RN0330, dopo la definizione diagnostica e rilascio di apposita scheda di malattia rara ad opera dei centri e/o presidi autorizzati dalla Regione.
Sia: “Ho la Sindrome di Ehlers-Danlos”
Nel 2019 l’artista australiana Sia ha dichiarato di essere affetta da questa condizione. Lo ha fatto su Twitter. E ha fatto bene, in quanto ci aiuta a capire che i social network possono essere uno strumento di sensibilizzazione molto importante per il mondo del sociale.
A quali associazioni puoi rivolgerti
Come ci conferma Gioia Di Biagio, uno dei problemi dell’Ehlers-Danlos è che “difficile riconoscere”. Inoltre, “al momento non c’è una cura”, ma vi sono diverse associazioni a cui è possibile rivolgersi, come:
- AISED, Associazione Italiana Sindrome di Ehlers-Danlos;
- A.Fa.D.O.C. , Associazione Famiglie di Soggetti con Deficit dell’Ormone della Crescita ed Altre Patologie.
Fonte prima foto: “Fragile”, di Ilaria Di Biagio